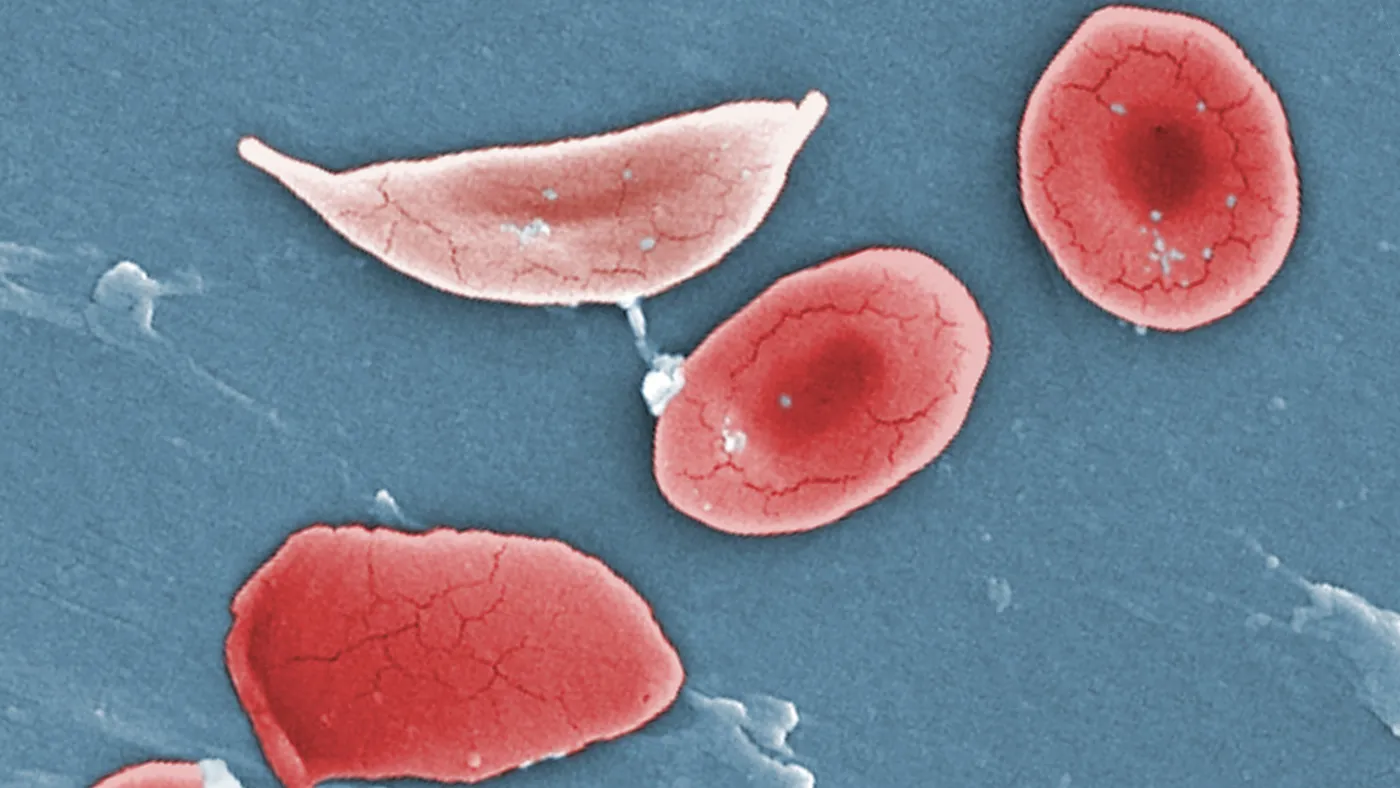
For most people, red blood cells travel smoothly through the body. Round and flexible, they flow through arteries and veins, carrying with them cargoes of life-sustaining oxygen.
But in a slice of the population, these cells are misshapen, contorted by a genetic mutation into sharp-edged scythes that snag in blood vessels. The blockages can cause acute agony — crises of pain that recur again and again in people with severe sickle cell disease.
These pain episodes have been a lifelong peril for Elinam Joe Tsogbe, putting him in the hospital more than a dozen times some years. The regular assault on his body has accumulated into deeper scars: a gallbladder removed at 14, a hip replaced at 19 and a dangerous trip to the intensive care unit in his early 30s.
Over the past three years, though, Tsogbe hasn’t had any pain crises.
In early 2021, Tsogbe received an experimental transplant of his own stem cells, which had been collected and edited in a laboratory using CRISPR gene editing, biomedicine’s most cutting-edge tool. Called exa-cel, the treatment is a kind of genetic workaround for sickle cell, built on decades of research into the disease’s roots. Among the dozens of people who, like Tsogbe, received it in clinical testing, the effects are life-changing.
“It was like day and night. I stopped having crises,” said Tsogbe, 37. “There was this newfound energy that was very different. You’re in disbelief, basically.” A DJ and a dancer, Tsogbe said that he can enjoy those activities for longer now and without the pain they previously caused.
Tsogbe’s disease hasn’t gone away. The mutation that causes sickle cell is still in his DNA, even if treatment with exa-cel has muted its most obvious and damaging consequences. The injuries done by the disease haven't been erased.
“Technically, we’re not cured. But clinically and physically, we are,” he said. “Now it’s not even apparent that we have this disease, because of everything we’re able to do.”
Changing the course of debilitating genetic diseases like sickle cell is the promise held out by CRISPR, and a main reason why the technology has spawned a growing pipeline of experimental drugs. Among them, exa-cel is the furthest along. Earlier this year, its developers, Vertex Pharmaceuticals and CRISPR Therapeutics, asked for approval from the Food and Drug Administration, which is set to decide by Dec. 8.
A clearance would be historic, coming some 10 years after scientists described CRISPR’s science-shaking potential in Nobel Prize-winning research. It would launch a new era of treatment for sickle cell, which most commonly affects people of African ancestry and was for decades overlooked by drugmakers. (Another genetic medicine that works in a different way could win an FDA OK a few weeks after the agency decides on exa-cel.)
Yet exa-cel’s benefits may not be accessible by all, or even most, of the tens of thousands of people estimated to have severe sickle cell in the U.S., never mind the millions more in other areas of the world where the disease is more prevalent.
The therapy is bespoke, created from an individual’s own stem cells via a laborious and expensive process. There are side effects, particularly during a preparatory chemotherapy regimen before exa-cel is infused. That regimen is so arduous that older people and those with organs damaged by sickle cell may not be healthy enough to receive it. The chemotherapy drugs can cause infertility, forcing a difficult decision on people who hope to have children.
Exa-cel also comes with the theoretical risk that wayward gene edits in the transplanted cells may one day lead to other problems, like cancer. Last week, a group of advisers convened by the FDA debated this possibility, but judged the risk to be small and exa-cel to be safe.
“Like most things in medicine, you’re trading other complications for a cure of the primary problem,” said Martin Steinberg, a hematologist and professor of medicine at Boston University who was involved in exa-cel research.
Medicines like exa-cel are therefore not an easy choice to weigh, even for people motivated to seek out new treatment options. Tsogbe said he spent a year researching the therapy and other experimental drugs before enrolling in Vertex and CRISPR Therapeutics’ trial.
Others may be similarly cautious, Tsogbe suggested: “A lot of people are skeptical about gene therapy.”
‘The lingering pain was gone’
Sickle cell is sometimes referred to as the world’s first molecular disease. Research in the late 1940s by chemist Linus Pauling and colleagues identified the disorder’s cause in molecular changes to hemoglobin, the essential oxygen-carrying protein found in red blood cells.
A typo in the gene that codes for one of hemoglobin’s principal parts deforms the protein in such a way that, when red blood cells release their oxygen, the hemoglobin binds together in rigid fibrils that warp the cell into a sickle. Now crescent shaped, the red blood cells pile up and block blood flow. These episodes are known clinically as vaso-occlusive crises.
“If blood [cells] in the muscles or the bones get stuck, patients have pain,” said Akshay Sharma, a pediatric hematologist at St. Jude Children’s Research Hospital who was an investigator in exa-cel’s trial. “If they get stuck in the brain, patients can have a stroke; if they get stuck in the heart, they can cause heart damage; if they get stuck in the lungs, individuals have acute chest syndrome.”
And when the blockages release, the sudden gush of blood into the choked-off tissue can also do damage. Recurring repeatedly over years, this stop-and-start flow of blood weakens organs, causing a constellation of symptoms that pervades daily life.
“The disease took a stronghold on my life and completely controlled what direction it went,” said Jimi Olaghere, 38, who was one of the first people treated in the exa-cel trial. “It progressively got worse as I grew older, with the constant pain that's the cornerstone of the disease. But it goes so much deeper than that,” he said. “My quality of life was really bad. At one point, I was just existing, basically trying not to die.”
There are a few drugs for sickle cell. One, called hydroxyurea and repurposed from its original use in cancer, can help some. But it doesn’t work well for everyone, has side effects and needs to be taken every day. Stem cell transplants from donors can be curative, but must be from someone whose cells have matching immune protein markers. Most people, including Olaghere, don’t have a match. And even for those who do, donor transplants carry significant risks, like graft-versus-host disease.
In searching for more effective treatments, researchers turned to a decades-old clue. Around the same time Pauling was probing the molecular basis of sickle cell, a pediatrician in New York named Janet Watson made an important observation. She noticed how infants with the disease didn’t show symptoms until months after birth, and proposed this was due to a fetal form of hemoglobin that’s produced in the first few months of life. This fetal hemoglobin then gives way to the adult hemoglobin that is mutated in people with sickle cell.
Later research found that certain gene variants can cause fetal hemoglobin to persist into adulthood. If this happens in someone with sickle cell, the disease is markedly less severe. (Hereditary persistence of fetal hemoglobin can also protect people from another blood disease called beta thalassemia.)
Finding a way to recapitulate this effect via a medicine has been a scientific holy grail for decades, said Stuart Orkin, a professor of pediatrics at Harvard Medical School who has spent his career studying blood disorders. “This entire time, it was pretty clear that fetal hemoglobin was protective for sickle cell disease and beta thalassemia.”
Exa-cel, which pairs CRISPR gene editing with stem cell transplantation, is meant to achieve that goal. To create the treatment, a patient’s stem cells are mobilized from the bone marrow into the blood, where they are collected and sent to a manufacturing facility. There, they are edited with CRISPR, which is used to cut a precise spot on a gene called BCL11A, and then shipped back to the treating hospital.
Once reinfused into the body, the edited stem cells mature into red blood cells that, thanks to the CRISPR snip to their DNA, contain fetal hemoglobin.
The process took the better part of a year for Olaghere. Cell collection alone stretched almost four months because his doctors weren’t able to extract enough of his stem cells on their first three attempts. After going through that and the chemo preparatory regimen, the actual infusion of exa-cel lasted only a few minutes, said Olaghere.
“When it was time for the infusion, they came into the room with three tiny syringes,” he said. “[It was] very strange, anticlimactic in a way. We had done all this work and here is all we had to show for it.”
Because his infusion took place early in the pandemic, Olaghere’s parents weren’t able to join him, his wife and young son in the room. They watched on Zoom as their son became one of the first few people in the U.S. to receive a CRISPR-based treatment.
Olaghere noticed exa-cel’s effects not long after. “I always had this lingering pain constantly, even when I took pain medication,” he said. “I remember the first noticeable difference after [being treated] was, two or three weeks later, I woke up and that lingering pain was gone.”
Today, the pain still remains dramatically less. Olaghere hasn’t had a crisis since receiving exa-cel and says he can do things, like ski, that would previously have been unimaginable. Now a father of three, he says he is relieved his children won’t have to worry about his health or about caring for him in the future. “I feel like I've never felt in my life before.”
His experience is shared by other participants in Vertex and CRISPR Therapeutics’ study of exa-cel, which has run for nearly five years and enrolled people with severe sickle cell between 12 and 35 years old. Among the 30 participants who have been followed for at least 16 months post treatment, all but one have remained free of pain crises for at least one full year, updated data included in FDA documents show. Twenty-seven haven’t had any pain crises at all.
Participants were able to stay out of the hospital and, in everyone treated with exa-cel, hemoglobin rose to levels considered to be protective from the disease’s symptoms.
“We clearly have reduced the disease severity quite substantially using this therapy,” said Haydar Frangoul, medical director of pediatric hematology at the Sarah Cannon Reserach Institute and a trial investigator.
“I think it proves, therapeutically, what we've known clinically all along,” said Steinberg, of Boston University. “If you have enough fetal hemoglobin broadly distributed throughout the red blood cells, then the consequences of sickle cell disease will melt away.”
The FDA and its advisers also appear convinced exa-cel works well. Recently published documents show agency scientists consider the therapy’s results to be “strongly positive” and a high-profile advisory committee meeting held Oct. 31 focused more on CRISPR technology rather than exa-cel itself. “We're not here discussing any concern with the benefit,” Nicole Verdun, head of the FDA office reviewing exa-cel, said at the meeting.
Is this a cure?
While exa-cel can eliminate pain crises, there is some reluctance to describe the therapy as a cure.
“A cure would mean all side effects associated with sickle cell disease are either halted or reversed, which we don’t know yet, and that such a change happens to be permanent and last lifelong, which again has not been shown yet,” said Sharma, of St. Jude’s.
It’s a debate that comes up often in discussions of genetic medicines, which in theory promise lasting benefit via a one-time treatment. But clinical trials of these types of medicines are relatively small and typically only run for a few years to obtain their principal results.
That’s the case with exa-cel, which has so far been given to 44 people in Vertex and CRISPR Therapeutics’ clinical trial. The first person treated in the trial, a 38-year-old woman named Victoria Gray, is now four years removed from her treatment. About 30 are between one and three years out — plenty of time to show exa-cel has made a dramatic difference, but not long enough to say that benefit will last the rest of their lives. The rest were treated less than a year ago.
“We need longer follow-up to really be sure that everything looks the way we hope it looks,” said Stephan Grupp, a transplant specialist at Children’s Hospital of Philadelphia and an exa-cel investigator. While he thinks the results to date indicate exa-cel’s benefit is durable, Grupp said five years’ worth of data would be more convincing.
One advantage of treating a blood disorder is that the effect of exa-cel can be easily monitored over time. Scientists generally think that fetal hemoglobin levels of 20% are enough to be protective from sickle cell symptoms. Data from the trial show treated participants’ levels remaining stable well above that threshold.
Vertex and CRISPR Therapeutics plan to track trial participants for 15 years. And if exa-cel wins approval, the companies intend to start a patient registry to do the same for at least some people treated commercially.
What exa-cel won’t do is fix injuries that sickle cell has already caused. “How well they do is how well they come to you,” said Frangoul, at Sarah Cannon. “I am not going to [be able to] resurrect a dead joint.”
Similarly, if repeated pain crises have damaged organs, or someone has previously had a stroke, exa-cel won’t reverse those effects. Doctors expect that preventing red blood cell sickling, as exa-cel does, will stop or slow further harm to organs, but exa-cel’s study has not run long enough to prove that, according to Grupp.
“Should we even be calling it a cure?” asked Teonna Woolford, head of the Sickle Cell Reproductive Health Education Directive, a patient group. “It means something different to so many people. Sickle cell is cumulative. If you already have this damage to your bones, having gene therapy is not going to fix that.”
Barriers to treatment
The FDA’s fast-approaching decision puts exa-cel on the horizon for people with sickle cell. But even if the agency grants an approval, treatment might not be as easy a choice as exa-cel’s benefits make it seem.
One of the most prominent concerns is the chemotherapy preconditioning that precedes treatment with exa-cel. Physicians use a chemo drug called busulfan that’s also standard for anyone who receives a stem cell transplant from a donor. This preparatory regimen is necessary to clear out a patient’s resident stem cells from the bone marrow so the newly edited cells can take root and multiply.
“If any of those stem cells are left behind, they can compete with the genetically modified cells over time and keep producing sickle cells,” said Sharma.
But like most chemo drugs, busulfan doesn’t discriminate. It attacks organs and other tissue, like the esophagus and mouth. It causes low blood cell counts, leaving patients vulnerable to infection during treatment. Older people may not be able to tolerate those side effects, according to Sharma.
Since Vertex and CRISPR Therapeutics capped the age of participants in their trial to 35 years, doctors weighing exa-cel for adults older than that won’t have representative data to support their decision, if the treatment is approved for wide use.
Most notably, busulfan can also lead to infertility.
“When I talk to a family about transplant, I talk about the risk of infertility, which is not like a theoretical risk,” said Grupp. “It’s a high risk of infertility.” (Sickle cell can cause infertility and other reproductive problems, too.)
This can force a wrenching trade-off for people seeking better sickle cell treatment so they can be healthy enough to have a family. Tesha Samuels, the president of the patient group Journey to ExSCellence and a person with sickle cell, had to confront this when she enrolled in a clinical trial of another gene therapy, developed by Bluebird bio, in 2017.
“For many years, I always wished for a treatment that would cure me of the disease so that I could become a mother,” said Samuels. “To then be given a chance at a cure at the risk of sacrificing the main thing you thought it would bring within grasp, was an overwhelming grief to overcome."
In testing, Vertex and Bluebird offered trial participants fertility preservation, such as by freezing and storing eggs. But while they covered that cost, pregnancy and birth via in vitro fertilization comes with its own set of expenses. And in the commercial setting, support may be less certain. (Vertex, in a statement, said it is “actively exploring options” and is “committed” to helping patients access such services.)
Woolford, who also has sickle cell, noted how the kind of guidance patients receive on fertility issues can vary widely outside of clinical trials. “We did some surveys and found that infertility was a huge barrier to these curative therapies.”
The price of treatment could be a barrier as well. Exa-cel is likely to cost several million dollars if the track record for other genetic medicines recently approved in the U.S. is any guide. Insurance companies are generally expected to cover it, given the high cost of care for people with sickle cell experiencing pain crises, but the upfront expense could still be large.
“Most healthcare systems are essentially set up to think about and pay for chronic medicines for chronic conditions,” said Stuart Arbuckle, Vertex’s chief operating officer, at a recent event hosted by BioPharma Dive. “Clearly these types of one-time treatments really challenge that paradigm.”
Vertex anticipates about 25,000 people in the U.S. and Europe will be eligible for exa-cel based on their disease severity. To help offset the financial hit, insurers may choose to link payment to patient outcomes. Arbuckle said Vertex plans to be flexible in exploring different payment models with different payers.
However, many people with sickle cell in the U.S. are covered by Medicaid, which is limited in its ability to test these types of contracts by budget and price reporting rules. A federal pilot program is planned, though it won’t start until 2026 at the earliest.
“Approval doesn’t necessarily mean access,” said Woolford. “That’s my concern: Who can actually access this?”

The genetic medicine era
Around a half dozen gene therapies for inherited diseases are already available in the U.S. If approved by Dec. 8, exa-cel would join their ranks as the first built around CRISPR gene editing, a technology barely a decade old. It would be a momentous milestone for science and for the biotechnology industry, emphasizing the arrival of a genetic medicine era.
Patients will face new questions in this era, when treatments can change their genes and potentially rid them of disease symptoms they’ve had for life.
Technologies like CRISPR will be an attraction for some. Olaghere said the gene editing aspect of exa-cel pushed him towards Vertex and CRISPR Therapeutics’ study, and that he had no trepidation about enrolling. “I'm a science fiction geek so I thought this was pretty cool.”
When weighing his options, Tsogbe decided he was comfortable with exa-cel because the treatment was built using his own cells. “It’s not anything else besides my own cells that are being edited and put back into me.”
Yet the dramatic benefits promised by exa-cel, and other genetic treatments like it, bring unexpected emotions, too. “You feel like you cheated,” said Tsogbe. “There’s a certain guilt that can affect you psychologically. You see other people who are going through exactly what you’ve gone through. Is it fair?”
Tsogbe and the others enrolled in the trials of exa-cel, Bluebird’s gene therapy and similar genetic medicines are some of the first people to confront these challenges. While the infrastructure to support treatment is being built, they’re pioneers in experiencing what comes after.
“I knew who I was and now I’m coming into a different identity,” said Samuels, who received the Bluebird gene therapy. “Who am I now without the illness? Where do I go?”
Samuels hasn’t had pain crises since her treatment. Her hemoglobin levels are normal. “I'm finding now that, if I go to a hematologist, they don't know what to do with me,” she said.
“In so many ways, we're blazing the trail,” Samuels added. “Hopefully so many more will be able to follow behind. That is my prayer and my hope.”




